Синдром Бругада (Brugada syndrome, BrS) — рідкісний, але потенційно летальний спадковий каналопатичний розлад, що підвищує ризик раптової серцевої смерті через шлуночкові аритмії. Цей синдром був вперше описаний братами Педро та Джосепом Бругада у 1992 році. Інформація подана, спираючись на сучасні дані з авторитетних джерел, таких як NCBI, PMC та Medscape.
Визначення
Синдром Бругада — це спадкова іонна каналопатія серця, що характеризується характерними змінами на електрокардіограмі (ЕКГ): неповна блокада правої ніжки пучка Гіса та елевація сегмента ST у правих прекардіальних відведеннях (V1–V3), без структурних змін серця. Це призводить до підвищеного ризику шлуночкової тахікардії/фібриляції (VT/VF) та раптової серцевої смерті (SCD). Синдром є рідкісним, з поширеністю 3–5 на 10 000 осіб, частішим у чоловіків (у 8–10 разів) і в азіатських популяціях.
Етиологія
Синдром Бругада є генетично детермінованим, з аутосомно-домінантним типом успадкування та неповною пенетрантністю. Найпоширеніша причина — мутації в гені SCN5A (15–30% випадків), що кодує альфа-субодиницю натрієвого каналу NaV1.5, призводячи до втрати функції. Інші гени включають GPD1L, CACNA1C, KCNE3, TRPM4 та понад 18 інших, що впливають на натрієві, калієві, кальцієві та HCN-канали. Тригери: лихоманка, електролітні порушення (гіперкаліємія, гіпокаліємія, гіперcalcемія), кокаїн, алкоголь, ліки (натрієві блокатори, трициклічні антидепресанти, ваготонічні агенти). Гендерна різниця з’являється після пубертату, можливо, через вплив тестостерону.
Патогенез
Патогенез включає порушення трансмембранних іонних струмів, переважно зменшення натрієвого струму (INa), що призводить до уповільнення провідності та дисперсії реполяризації. Існують три основні гіпотези:
- Реполяризаційна гіпотеза: Збільшення транзієнтного вихідного калієвого струму (Ito) в епікарді правого шлуночка створює градієнт потенціалу, що викликає елевацію ST та фазу 2 реентрі.
- Деполяризаційна гіпотеза: Мутації SCN5A уповільнюють деполяризацію в правому шлуночковому відтоку (RVOT), з фіброзом та зменшенням щілинних з’єднань (конексин-43), сприяючи реентрі та хвильовим перервам.
- Гіпотеза невідповідності струму-навантаження: Структурні зміни (фіброз у RVOT) викликають збудження з невдачею через зменшення внутрішніх струмів.
Аритмії часто виникають уночі через підвищений вагальний тонус.
Клінічні прояви
Симптоми варіюють від асимптоматичних (72% пацієнтів) до раптової смерті. Основні: синкопе (80% у пацієнтів з VT/VF), запаморочення, серцебиття, нічні кошмари, агональне дихання. Раптова смерть часто трапляється під час сну або відпочинку, без продрому. Асоційовані: атріальна фібриляція (10–30%), суправентрикулярна тахікардія, синдром хворого синуса. Тригери: лихоманка, великі прийоми їжі. У 28% немає сімейного анамнезу SCD.
Діагностика
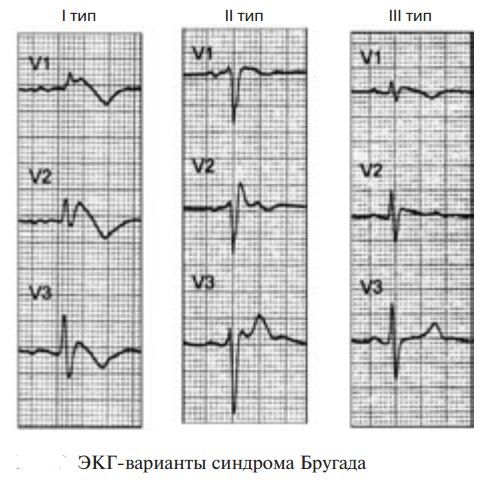
Діагностика базується на ЕКГ:
При синдромі Бругада на ЕКГ (тип 1, діагностично значущий):
- Псевдоблокада правої ніжки пучка Гіса – комплекс QRS у V1–V2 нагадує RBBB, але з коротшою тривалістю.
- Підйом сегмента ST у V1–V3 ≥2 мм, куполоподібний або сідлоподібний.
- Інверсія зубця T у V1–V3 після підйому ST.
- Відсутність структурних змін серця (за даними ЕхоКГ або МРТ).
- Типи ЕКГ:
- Тип 1 — куполоподібний підйом ST ≥2 мм у ≥1 з відведень V1–V3 з інверсією T.
- Тип 2 — сідлоподібний підйом ST ≥2 мм, позитивний або двофазний T.
- Тип 3 — куполоподібний або сідлоподібний підйом ST <2 мм.
Тип 2/3 (сідлоподібні) — підозрілі, вимагають провокації. Провокаційний тест: введення блокаторів натрієвих каналів (флекаїнід, аймалін) для розкриття патерну. Додаткові: генетичний тест на SCN5A, електрофізіологічне дослідження (індукція аритмій), ЕхоКГ/MRI для виключення структурних змін, лабораторні тести (електроліти, тропонін). Диференціальна: Brugada phenocopy (від електролітів, ішемії), АРВД.
Лікування
Основне — імплантація кардіовертера-дефібрилятора (ICD) для пацієнтів з абортованою SCD, синкопе або спонтанним типом 1 ЕКГ. Фармакотерапія: хінідин (блокує Ito, нормалізує ЕКГ), бепрідил, цілостазол для VF-штормів. Абляція: радіочастотна абляція RVOT, особливо епікардіальна, перспективна. Асимптоматичні: моніторинг, уникнення тригерів. Генетичний скринінг родичів.
Прогноз
Синдром відповідає за 4% усіх SCD і 20% у структурно нормальних серцях; середній вік смерті — 41 рік. Ризик: вищий у чоловіків, з синкопе, спонтанним типом 1, індукованими аритміями. ICD знижує смертність, але ускладнення (неадекватні шоки) — 8,2% подій за 24 місяці. Раннє втручання покращує виживання; генетичне консультування обов’язкове.
У висновку, синдром Бругада вимагає мультидисциплінарного підходу з акцентом на ризик-стратифікацію та профілактику.